
Welcome!
recently published
organic semiconductors
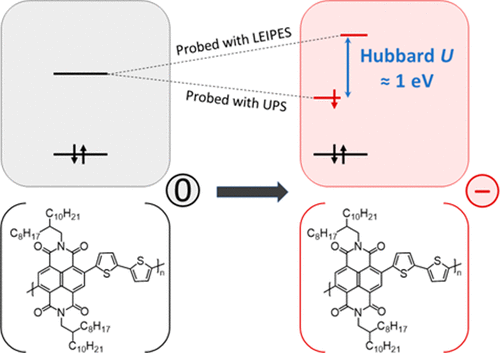
Spectral signatures of a negative polaron in a doped polymer semiconductor: Energy levels and hubbard U interactions
D. Lungwitz et al. | J. Phys. Chem. Lett. 14 (2023)
The modern picture of negative charge carriers on conjugated polymers invokes the formation of a singly occupied (spin-up/spin-down) level within the polymer gap and a corresponding unoccupied level above the polymer conduction band edge. The energy splitting between these sublevels is related to...
perovskites
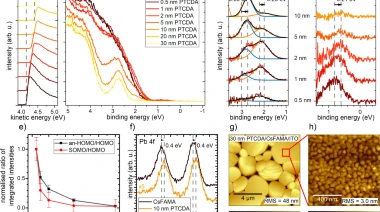
Effect of ground state charge transfer and photoinduced charge separation on the energy level alignment at metal halide perovskite/organic charge transport layer interfaces
L. Frohloff et al. | Appl. Phys. A 129 (2023)
One of the most promising routes for the future fabrication of solution-processable high-performance solar cells is to employ metal halide perovskites as photoactive material combined with organic semiconductors as charge extraction layers. An essential requirement to obtain high device performance...
2D semiconductors
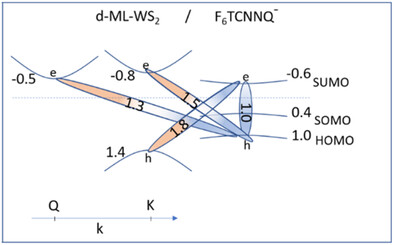
Defect-dependent optoelectronic properties at a molecular p-dopant / monolayer WS2 interface
J. Ma et al. | Phys. Status Solidi A (2023)
Photoemission and optical absorption experiments indicate that native n-type defects of monolayer WS2 lead to interfacial negatively charged molecular acceptors, which support interlayer charge-transfer excitons.
News
Lise-Meitner-Prize for Dr. Dominique Lungwitz
Dominqiue Lungwitz received a Lise-Meitner-Prize 2022 for her dissertation. Congratulations! More about the prize and "Vereinigung der Freunde und Förderer…
The IOP – Humboldt Postdoctoral Fellowship in Physics
Nominations are open for postdoctoral fellowships between two cities, Berlin and Beijing, as part of a joint physics program between…
New doctoral program “Ciencia y Tecnología de Materiales” at Pontificia Universidad Javeriana
Two speakers from Berlin (HU and HZB) at the inaugural symposium of the new doctoral program “Ciencia y Tecnología de…
Integrative Research Institute for the Sciences

Helmholtz-Zentrum Berlin
Department of Physics

Humboldt-Universität zu Berlin
